Evidence for the Evolutionary Model
Subtitle
ERVs
Three Layers of Endogenous Retroviral Evidence for the Evolutionary Model
[Electronically published: November, 2009; Last Updated: May 2012]

Contents (jump to section by clicking; return to top using your browser's backspace button)
Introduction
A powerful source of evidence that modern species diverged from ancestral species via descent with modification is that of endogenous retroviruses (ERVs). ERV evidence consists of three independent layers that corroborate one another. As will be further discussed, the three layers of ERV evidence are: 1) the sharing of ERVs in identical loci among organisms of varying degrees of taxonomic separation, and the nested hierarchies that these shared ERVs are arranged in; 2) the examination of shared mutagenic discrepancies between shared ERVs, so as to infer relative sequence of insertion; and 3) the nested hierarchies of shared mutations among given ERVs in identical loci. But before this evidence can be examined, one must have a firm understanding of retroviruses and how they infect cells.
Life-cycle and Ancestral Implications
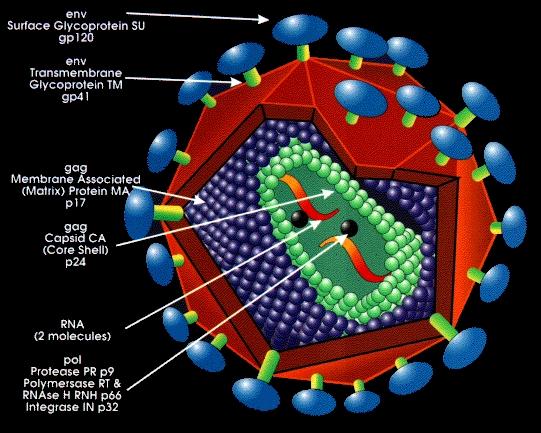
In order to replicate itself, a retrovirus needs to use the molecular machinery of a host, and it begins the process by first binding its extracellular and transmembrane glycoproteins to a cell’s coreceptors. The capsid—containing the retroviral enzymes reverse transcriptase, integrase, and protease, as well as two copies of the retroviral genome—is inserted into the cell’s cytoplasm, where it uncoats. Now in its host cell, a retrovirus reverse-transcribes its genome from RNA to DNA via reverse transcriptase. Protease then process the viral DNA by removing a dinucleotide off each 3’ end, and integrase inserts it in the host cell’s genome (Targeting HIV replication, n.d.). Once integrated, and in DNA form, its genome is known as a prototype retrovirus, or provirus. Upon integration, the cell is allowed to divide, and eventually the presence of certain environmental conditions trigger proviral activation. Copy after copy of the retrovirus is produced as virions bud off, mature, and go on to infect other cells, leading to the death of the infected cell.

The would-be problem for the retrovirus is that once the cell replicates and activation occurs, the provirus relies on the RNA polymerase II of the host cell to transcribe it into viral RNA to be packed into its virions. Unfortunately, RNA polymerase II is designed to transcribe messenger RNA (mRNA) for translation into polypeptides, so although it uses promoters to initiate transcription, since they don’t code for amino acids, it doesn’t transcribe them. With only one set of promoters on the proviruses, the newly transcribed retroviral genomes would have no promoters at all. When the virions that contained them would reverse transcribe and insert them into new host cells, the second round of activation and transcription would never be able to occur.
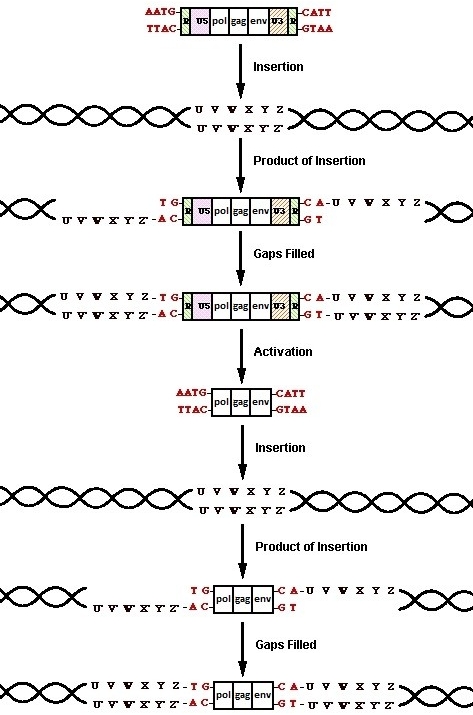
Retroviruses circumvent the problem of vanishing promoters by simply polymerizing copies of them during reverse transcription. They achieve this by possessing identical sections of DNA, called repeats, on either terminus of their genome. Early in the process of reverse transcription, the first jump occurs, in which the transfer RNA (tRNA) primer detaches and the DNA repeat hybridizes with the remaining RNA repeat at the genome’s 3’ terminus (Cann, n.d.). Given the relatively small size of the repeats, if they are not identical, they cannot hybridize. As for the 5’ unique (U5) and 3’ unique (U3) sections, a copy of each is polymerized on the opposite terminus. Between the necessity of identical repeats, and the duplication of unique sections, the resulting U3-R-U5 sections, called long terminal repeats (LTRs), must likewise be identical at the time of insertion. This will become very important later on in examining the second and third layers of ERV evidence.
Once the provirus has been processed, integrase begins the process of insertion by breaking two of the host genome’s phosphodiester bonds between a 3’-hydroxyl group and 5’-phoshphate group, allowing the provirus to insert in a highly randomized manor (Skinner et.al., 2001). Different retroviral integrases have minor statistical biases for insertion within the general area of “chromosomal regions rich in expressed genes… [their interwoven] CpG islands… active genes… [and areas] near transcription start sites (Mitchell et al., 2004),” but these biases are so minor, that they require thousands of trials (3127 used in the Mitchell study) just to detect them. For the most part, the process of integration is observed to be quite random.

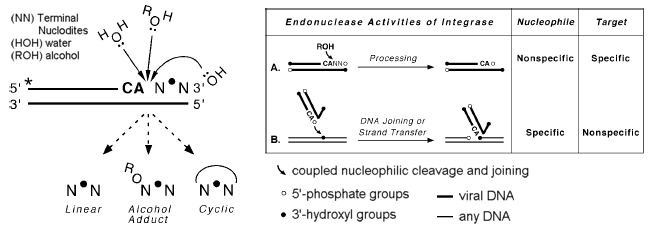
The two phosphodiester bonds integrase breaks are on either strand of the host DNA. Rather than breaking the bonds between two base pairs, integrase separates the breaks by several base pairs, forming a jagged cut (Skinner et al., 2001). Since each of the two strands of DNA are complimentary to one another, the overhangs on either end of the jagged cut are likewise complementary. When nucleotides are polymerized to fill the gaps, the end result is two identical stretches of the DNA between the two initial breaks flanking the inserted provirus. This effect is called target site duplication. The second effect of the insertion is the displacement of DNA. This can be observed by comparing corresponding genomic areas in species with and without the provirus. The ones with the provirus display a target site duplication, where as the ones without the provirus display the preintegration site (Polavarapu, Bowen, & McDonald, 2006). The presence of these two effects conclusively demonstrates that the sequence in question was inserted into the host's genome via a transposase enzyme at some point in history of the host's lineage.

The targets of retroviruses are usually somatic cells, but if the infected cell happens to be a sperm or egg cell, known as a gamete, or a testicular or ovarian cell that divides into a gamete, that gamete may be used to produce an offspring. In such a case, the provirus becomes a permanent fixture within the offspring’s genome. Its permanence is due to fact that “there is no mechanism for removing proviruses precisely from the genome, without leaving behind a solo LTR or deleting chromosomal DNA (Johnson and Coffin, 1999).” Although the retrovirus was foreign to the organism it infected, and thus would be considered exogenous to that organism, once passed on to the organisms offspring, it would be present in the offspring’s natural, healthy state, and thus would considered endogenous to it.
Now being passed on from one generation to the next, ERVs accumulate copying mistakes by DNA polymerase during subsequent host cell replication. Since ERVs are generally not conserved, they accumulate mutations at the same rate as introns. And, as with introns, over time, the mutations can become fixed in the host population’s gene pool (Boeke and Stoye, 1997. In Coffin, Hughes, & Varmus, 1997). Given enough time, enough mutations accumulate to render the ERV incapable of activation.
If two or more individuals have ERVs of the same family at the same loci, one might think that there exist two plausible explanations: 1) that a retrovirus inserted in a common ancestor, and was passed on to the individuals via sexual reproduction; or 2) that separate retroviruses inserted in the same loci in separate ancestors, and were passed on to the individuals from each respective ancestor. What rules out the latter of these explanations for the majority of shared ERVs is the highly random nature of integration discussed earlier, and shared mutation among ERVs in identical loci, to be discussed shortly.
Thus, we can then conclude—regardless of how many individuals have retroviruses in the same loci of their genomes—the majority of those retrovirus necessarily inserted within the genomes individual cells of individual ancestral organisms common to each of them, and were passed on to those individuals via sexual reproduction. If the organisms are of the same species the individual from which the insertion originated may likely have also been of the same species. If, however, the organisms are of different species, the shared ERVs are referred to as orthologous, and that is when things get interesting.
The Three Layers of ERV Evidence for Common Ancestry
Layer 1
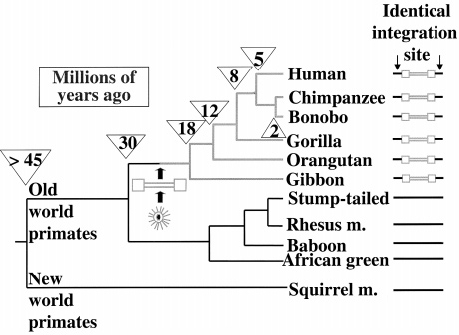
When we examine the collective genome of Homo sapiens, we find that a portion of it consists of ERVs (IHGS Consortium, 2001). We also find that humans share most of them with Chimpanzees, as well as the other members of Hominidae (great apes), the members of Hylobatidae (gibbons), and even the members of Cercopitheciodae (old world monkeys) (Kurdyukov et al., 2001; Lebedev et al., 2000). Since humans don't and/or can’t regularly procreate and have fertile offspring with members of these species, and thus don’t make sizable contributions to their gene pools, and vice versa, their inheritance cannot have resulted from unions of modern species. As previously mentioned, parallel integration is ruled out as an explanation for the many shared ERVs by the highly random target selection of integrase. And even if it was far more target-specific than observed, it would require so many simultaneous insertion and endogenizations that the evolutionary model would still be tremendously more parsimonious. This leaves only one way the majority of these ERVs could have been inherited: via sexual reproduction of organisms of a species that later diverged into the ones the organisms that share the ERVs belong to, i.e. ancestral species—simply put, most ERVs are orthologous; humans and the other primates must share common ancestry.

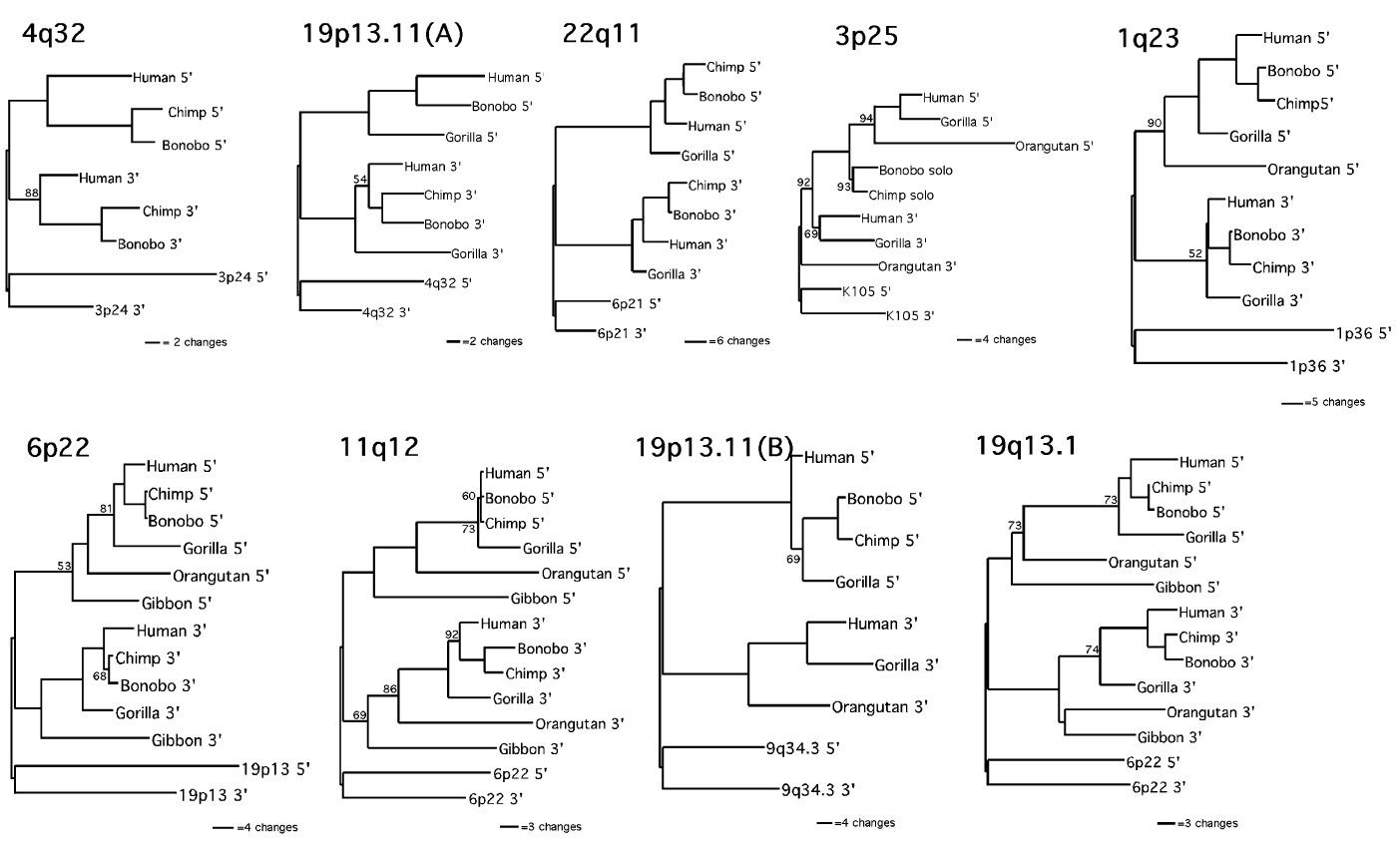
Not only are there many ERVs shared among primates, but they are shared in hierarchical subsets of the whole. Each set falls within another set, giving an unbroken line of inheritance for every species (Kurdyukov et al., 2001; Lebedev et al., 2000). This pattern is called a nested hierarchy. These patterns further corroborate that the many species of primates share common ancestry, and necessitate a specific sequence of divergence from one ancestral species to the next. They are wholly inexplicable by the model of uncommon ancestry.

Layer 2
As previously explained, although the LTRs of a provirus must be identical upon insertion, once endogenized, they begin accumulating mutations. Any mutations to one LTR become quite apparent, as they are not accompanied by the same mutations in the other. Thus each mutation causes the ratio of discontinuity between the two LTRs of a full-length ERV to increase. Since ERVs in identical loci among greater numbers species of wider taxonomic separation correlate to older insertions, if the evolutionary model is correct, they should also have higher ratios of discontinuity between their LTRs. And what do we find? We find just that; a pattern, where the degree of a shared ERVs’ LTR-LTR discontinuity is proportional to the degree of taxonomic separation between the species that share it (Johnson and Coffin, 1999). There is deviation from the pattern—likely caused by viral transfer and interelement recombination/conversion (Hughes & Coffin, 2005) and viral transfer (Belshaw et al., 2004)—but the pattern is holds for many full-length ERVs and is explainable only by decent with modification from a specific series of common ancestral species. Once again, we see strong evidence for ERV orthology.
Layer 3
When the mutations in shared ERVs are examined, many are found to be identical to others. Just as will the distribution of ERVs, some shared mutations within a single shared ERV fall into nested hierarchies; some are shared by all, many by subsets of the whole, and each set falls within another set (Hughes & Coffin, 2005; Johnson and Coffin, 1999). Despite deviation caused by the same mechanisms effecting LTR-LTR discontinuity ratios, many of these nested hierarchies of mutation match those of distribution. Part of what makes this such powerful evidence for the evolutionary model is that is that ERV distribution and mutation rely on entirely different mechanisms; the function of integrase and the DNA replication complex, respectively. That the two nested hierarchies match at all is only explicable by common ancestry. The evidence for ERV ortholgoy is clear.
Summary
The three layers of ERV evidence that have just been laid out are as follows:
Layer 1: the presence of ERVs in identical loci among species of various degrees of taxonomic separation, and of the nested hierarchies they fall into.
Since they’re passed on through sexual reproduction, the many ERVs fixed in identical loci in different species necessitates the past presence of a species ancestral to both, that has since diverged into the two modern ones. And the patterns of their distribution indicate a specific sequence of divergence.
Layer 2: the comparative degrees of LTR-LTR discontinuity among full-length ERVs in identical loci.
Since LTRs are identical upon reverse transcription and subsequent insertion, greater divergence correlates to an older insertion. Thus the patterns of discontinuity indicate sequences of divergences consistent with those indicated by distribution.
Layer 3: shared mutations among ERVs identical loci and the corresponding nested hierarchies they fall into.
Since mutations accumulate and fix in populations of organisms, the distribution of shared mutation indicate a sequence of speciation events consistent with that which is indicted by both distribution and LTR-LTR discontinuity.
The majority of ERVs really did originate in a common ancestral species; most truly are orthologous.
Amount of Shared ERVs
With so many claiming that it's only around 7 or 14, an important question is; just how many ERVs do humans share with chimpanzees? The answer is that humans and chimpanzees share virtually all of them. We know this is the case for two reasons; examination of indel variation, and whole-genome analysis.
Total Indel Variation
When lining up genomic sequences for comparison, there are many ways to measure difference. The most common is the measurement of substitutions; a base pair differing from one sequence to the next, such as an A, instead of a G. But an equally important measurement is that of indels:

An indel is a deletion or insertion of a sequence in an organism’s genome. When the genomes of multiple organisms are aligned, an indel in either genome will result in a gap. This is useful in determining the how many ERVs, Alus, or any other types of transposons are shared, since insertions at a given locus in only one lineage or only the other will result in gaps, yet insertions in identical loci leave no gaps:
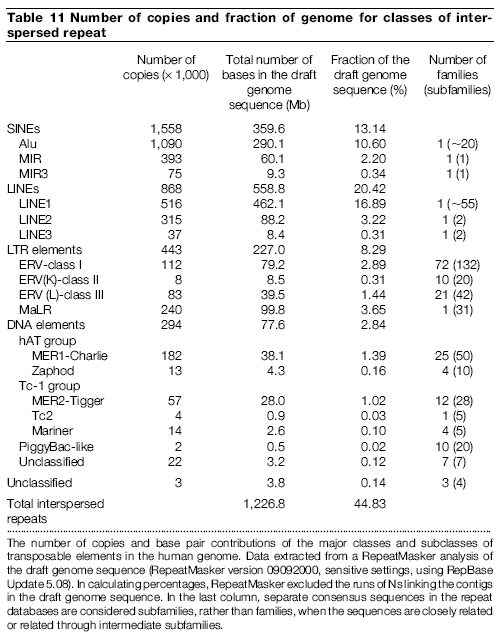

The total length of all ~6.7 million transposable elements in the human genome is at least ~1.2 Gb (gigabases; billion base pairs), and the total length of all ~200 thousand ERVs is at least ~127 Mb (megabases; million base pairs) (International Human Genome Sequencing Consortium, 2001). But the total indel variation between the chimpanzee and human genomes is only ~3%, comprising a maximum of ~45 Mb (~1.5%) in each genome (Chimpanzee Sequencing and Analysis Consortium, 2005). Remember; that includes deletions and duplications, as well as the insertion of transposable elements, like ERVs. So only a fraction the ~45 Mb in the human genome could even potentially be ERVs in different loci. Even if every single ERV-sized indel in the human genome was an ERV at different loci between chimpanzees and humans, that would only represent a small fraction of the ~127 Mb of all ERVs; and a minute fraction of the ~1.2 Gb of all transposable elements. Thus, right from the start, we know that the majority of ERV are in identical loci.
Indel Variation Observed to Involve ERVs
Total indel variation provides a minimum number of transposable elements that can be shared in identical loci between chimpanzees and humans; but further examination is necessary to determine the actual number. One way this can be done is by isolating only the indels that are the right size to potentially be ERVs in different loci. Once this is done, the sequences corresponding to those gaps can be individually examined.
The results of such analysis are that less than 100 ERVs are human-specific (Polavarapu, Bowen, & McDonald, 2006). As previously stated, if a sequence is not only at a given locus in one lineage, nor only at a given locus in the other, then the only remaining possible state in which it can exist is at the same locus in both lineages. So the number of ERVs in identical loci is the total number minus the number that form gaps. With less than 100 of the ~200 thousand ERVs in the human genome yielding no gaps, the percentage of ERVs in identical loci is grater than 99.9%.
Whole-genome Analysis
In 2005, the available sequence of the Chimpanzee genome was aligned with that of the human genome, and an extensive comparison analysis was performed. As part of this analysis, the researchers looked at every available solo LTR and full-length ERV in the chimpanzee genome, and checked to see if there was also one at each corresponding locus. Just as with the examination of indels, the results were that less than 100 ERVs are human-specific and less than 300 ERVs are chimpanzee-specific (Chimpanzee Sequencing and Analysis Consortium, 2005; R. Waterston, personal communication, April 22, 2010).

In summary, indel variation shows that most transposable elements, such as ERVs, cannot be lineage-specific; they must be in identical loci. When the indels are examined, this is corroborated, and less than 0.1% of ERVs are found to be lineage-specific. Finally, definitive confirmation is obtained by genome-wide comparison, where virtually all ERVs are directly observed to be in identical loci.
ERV Predictions from the Evolutionary Model
As primate genomes continue to be sequenced in full, and compared to the human genome, it will be found that as the degree of taxonomic separation of the compared lineages increase:
1) The ratios of ERVs, Alus, and other transposable elements not in identical loci to those in identical loci will gradually increase—from the current human to chimpanzee ratios of ~0.1% for ERVs and ~0.6% for Alus (IHGS Consortium, 2001; CSA Consortium, 2005).
Explanation: The older the divergence between the compared lineages, the more time each has had as a distinct lineage; thus the more independent insertions each should have accumulated.
2) The solo-LTR to full-length element ratio of the ERVs in identical loci will rapidly increase.
Explanation: Insertions tend to rapidly undergo homologous recombination, but as soon as it begins accumulating mutations, its chance of recombination rapidly decreases (Belshaw et al., 2006). Thus there are very old full-length ERVs, but their numbers fall off quickly with age.
3) Transposable elements in identical loci will largely be arranged in accordance with the current nested hierarchy of examined elements, but between insertion symplesiomorphy caused by incomplete lineage sorting (i.e. allelic segregation), and insertion homoplasy caused by target site preference, the amount of pattern deviation will increase beyond the current solitary case of HERV-K-GC1. The majority of this increase will involve the deviation of only one lineage per deviant identically positioned ERV.
Explanation: A site being used twice is quite rare, and a site being used three times is not observed; even in a sample of 40,528 HIV insertions (Wang et al., 2007). This—in addition to tendency towards allelic fixation—indicates that insertion symplesiomorphy/homoplasy will likely be limited to only one point of deviation at a time; as is the case with HERV-K-GC1 (Barbulescu et al., 2001) and Ya5AH137 (Salem et al., 2003).

Common Creationist Responses
There are three common responses to ERV evidence by Creationists:
a) The first is to ignore most of it—most notably of which being the patterns of distribution and mutation.
Creationists often directly respond to ERVs in identical loci by arguing that integrase can generate them when inserting in separate lineages, and indirectly respond to the hierarchical grouping of such ERVs by citing HERV-K-GC1 and (incorrectly) citing CERVs of families 1, 2, and 3.
But they do not appear to respond to the positive correlation between number of species that share ERVs in identical loci and the discontinuity between the 5’ and 3’ LTRs of those ERVs (Johnson and Coffin, 1999); nor to the sharing of mutations between the LTRs of ERVs in identical loci, the hierarchical grouping of such mutations, or that the nested hierarchies of distribution match those of mutation (Hughes & Coffin, 2005).
It remains true that common ancestry is a powerful predictive model with regards to the placement of ERVs. It predicts that most ERVs should be shared in certain hierarchical sets and not in others, and so far, almost every one has been where predicted.
A comprehensive working creationist model that is consistent with uncommon ancestry and incorporates the whole of ERV data would be an important next step, but has thus far gone unformulated.
b) The second common response is to use deviation from these patterns they fail to address as justification for dismissing ERVs outright. The following represents the two most common examples:
HERV-K-GC1
Although patterns are formed and act as powerful evidence of past occurrences and of mechanisms that causes them, the complex nature of the physical world (due to so many simultaneous interactions on so many levels) often causes deviation from those patterns.
Thus the patterns act as indicators of what is actually going on, and deviation from them is both expected and rendered likely not indicative of what is actually going on.
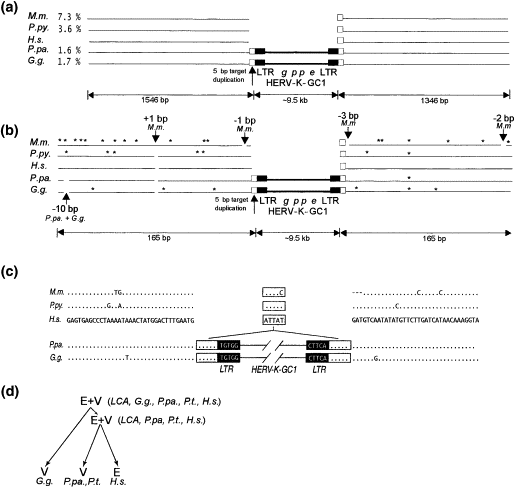
There is only one, solitary known deviation of the distributional nested hierarchy; a relatively recently endogenized/fixed ERV called HERV-K-GC1. Since this is an unrepresentative deviation from a pattern, all that needs to be done is identify mechanisms that can account for that deviation. There are at least two known mechanisms to account for it (Barbulescu et al., 2001):
1) The insertion was in an allele that remained heterozygous in the populations across two divergences, given genetic drift and the divergent sub-population size (allelic segregation).
More detailed explanation:
When a provirus inserts, it does so in only one copy of the genome, and when it becomes endogenized, it is heterozygous in the organism. Both the allele with the integration and the preintegration site exist in the population. Since the ERVs are not conserved, they can persist in the populations, but since the sexually separated subpopulations from which a new species would diverge can easily be very small, and since genetic drift is quite prevalent in small populations, alleles can be lost, making the other fixed in the population.
HERV-K-GC1 would have inserted and endogenized in the common ancestor of humans, chimps and gorillas, but would have been represented well enough in the split subpopulation that went on to later diverge into the ancestor of humans and chimps that the allele remained. In the split subpopulation that went on to later diverge into the ancestor of humans, the allele would have been fixed by genetic drift.
2) The insertion was in a duplicate section of the chromosome that underwent homologous recombination in each respective linage.
More detailed explanation:
The other possibility is that HERV-K-GC1 inserted in a duplicate section of the chromosome in the ancestor of humans, chimps and gorillas, (possibly soon after the split form the ancestor of orangutans) and became fixed, likely soon after that, but possibly in any order. That fixed allele then underwent homologous recombination in each respective linage.
The distributional nested hierarchies are clear, and match those of shared mutations. Both of those patterns are also corroborated by those of long terminal repeat discontinuity. Deviation from patterns is expected, but the presence of these patterns is powerful evidence of common ancestry.
Family 1, 2, and 3 CERVs
Insertions of CERV families 1, 2, and 3 are found in the genomes of gorillas and chimpanzees, but not in human genomes. What must be understood is that only nested hierarchies formed by the inheritance of orthologous ERVs provide evidence for common ancestry; not non-orthologous ones, since they are necessarily integrated in separate ancestors.
Each of the examined PtERVs claimed to be violating the nested hierarchy are in non-orthologous loci—they simply have uncharacteristically high long terminal repeat discontinuity ratios, likely due to interelement recombination/conversion and viral transfer (Polavarapu, Bowen, & McDonald, 2006).
As stated in a publication on PtERV1, by Yohn (2005, p. 578-579) and his colleges, "275 (95.8%) of the insertion sites mapped unambiguously to non-orthologous locations."
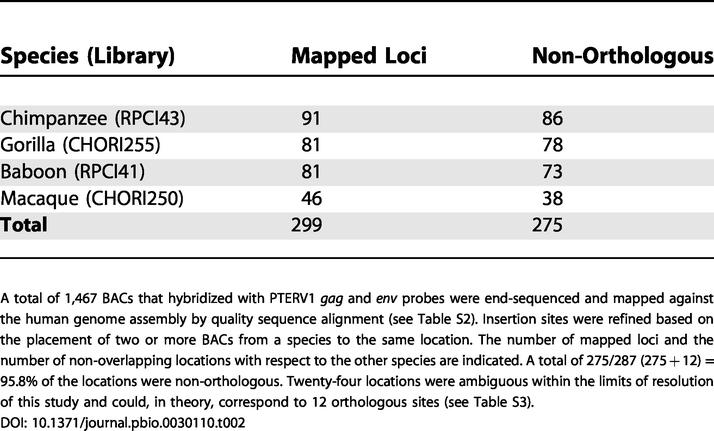
The remaining 24 (4.2%) insertions "could not be definitively resolved as orthologous or non-orthologous" due to the limitations of the "BAC-based end-sequencing mapping approach" used. Part of the results was derived by comparing "three intervals putatively shared between macaque and chimpanzee" to "the available whole-genome shotgun sequences for [the] two genomes."
Upon so doing, there were two instances where they "were able to refine the map location to single basepair resolution... Although the status of the remaining overlapping sites is unknown, [the data resolved] four additional sites as independent insertion events and suggest that the remainder may similarly be non-orthologous."
All this indicates that these insertions of these three families of ERVs infected primates after chimpanzees and humans diverged. Humans happened to either escaped infection due to geography or immunity, or were infected but none were ever endogenized and/or fixed in the population. They are not examples of deviation from the nested hierarchy of ERV distribution.
c) The third common response is to use red herrings to dismiss ERV evidence. The two examples discussed here are target site preference and functionally:
Target Site Preference
It is true that the insertion of integrase is not entirely random, but it is nowhere near locus specific.
Figure 1 on page 1129 of the Mitchell study (2004) shows that the 3127 proviruses analyzed inserted in different loci. The study merely demonstrates the high order preference for the general areas of "chromosomal regions rich in expressed genes... CpG islands... active genes... [and areas] near transcription start sites."

It is the same with the Taruscio (1991) study, as it clearly sates:
"Retroviruses have the ability to integrate into the genome of their host, in many cases with little apparent sequence or site specificity. However, relatively few studies have addressed more general features of chromosomal integration... These investigators have suggested that higher order structural features, or an active transcriptional capacity of chromatin may be fundamental for preferred integration events (p.141-142)."
It is clear from all studies on target site preference that while proviral insertion is not purely random, it is also not locus specific; due to the way it directly attacks the 5' and 3' phosphodiester bonds, with no need to ligate (Skinner et al., 2001). So relative to pure randomness, insertion is non-random, but relative to locus specificity, insertion is highly random.
Since the model of uncommon ancestry requires near locus specificity, bringing up relatively small biases for "higher order structural features" is a red herring.
Functionality
There is no question that some ERVs have functions in organisms, but there are no wholly functional ERVs—only functional components, with the remainder deleted or mutated into non-functionality.
For instance, the contribution of enJS56A1 and enJS5F16 (of the mere ~20 enJSRVs) to placental growth/differentiation regulation is achieved solely by their env genes with open reading frames (Dunlap et al., 2006; Palmarini et al., 2000). Although they also have an open gag reading frame (causing gag-gag interaction that restricts pathogenic JSRVs), they are the only ones known to have this (Mura et al., 2004). And every studied enJSRV has a closed pol reading frame (Murcia, Arnaud, & Palmarini, 2007).
Another example is the transcriptional contribution of LTRs to genes' promoters:
1) Not only are most ERVs not at a loci that even makes it possible for them to contribute to transcriptional activity, but most ERVs have recombined into solo LTRs. Since only the LTRs of active full-length ERVs can contribute (Cohen, Lock, & Mager, 2009, p.107), even most ERVs in the right position have no effect. Just as with enJSRVs, these ERVs represent a very small percentage of the whole.
2) The actual genes of these ERVs contribute nothing—only their promoter sequence-rich LTRs do. Again, just as with enJSRVs, these are examples of functional ERV components, rather than functional ERVs.

It is the same with every case observed; again, there are no "functional ERVs;" only a small percentage of ERVs with functional components.
But it's a moot point, because we know that ERVs are insertions:
The hallmark of an insertion is a displacement of chromosomal DNA, and the hallmark of insertion by integrase is the presence of target site duplication, due to the way it attacks the 5' and 3' phosphodiester bonds with an offset of a few base pairs (Skinner et al., 2001). Since full-length ERVs are accompanied by target site duplications and DNA displacement, they are necessarily endogenized/fixed proviral insertions.
So any functional components are necessarily post-insertion exaptations, and the fact that they are necessarily insertion means that they can not be part of any 'original design.' The issue of functionality is simply a red herring, when discussing how ERVs necessitate common ancestry.
For an understanding of scaffolding (p.365-366) and exaptation (p.361-363), including various "paths to exaptation," refer to the first half (p.358-366) of "The Evolution of Complex Organs" by Dr. Gregory (2008).
d) The fourth common response is to understate the number of ERVs in identical loci, and to use that number in conjunction with target site preference. The argument is as follows:
There are only a few ERVs found in identical loci in both humans and chimpanzees. Given target site preference, it is not unlikely that they are the result of infections in separate lineages. Thus the sharing of ERVs is constant with uncommon ancestry.
It is not unreasonable to hypothesize that among the tens of thousands of ERVs, similar target site preference may have resulted in a very small percentage of instances of insertion, endogenization, and fixation in identical loci in two separate lineages.
But the first problem with the idea that this renders uncommon ancestry plausible is that there are ERVs shared by many more than just two lineages. For instance, there are ERVs shared by chimpanzees, humans, gorillas, orangutans, gibbons, and Old World Monkeys (Kurdyukov et al., 2001; Lebedev et al., 2000). The second problem—and by far, the most important—is that we do not share only a few ERVs in identical loci with Chimpanzees; examination of indel variation, and whole-genome analysis shows that we share virtually all of them with Chimpanzees. This is discussed extensively in the "Amount of Shared ERVs" section, above.
Conclusion
Ultimately, the best way to respond to such claims—after having addressed their points specifically, of course—is to relentlessly drive home what they seem least willing to discuss; that deviation from patterns is to be expected, and that the corroboratory patterns of distribution, mutation, and LTR-LTR discontinuity are solely explicable by the evolutionary model.
Search This Site:
Google: Yahoo: MSN:
Modifications and corrections are frequent. All information is subject to change.
No comments:
Post a Comment