My comments rendered in red.

The Natural History of Retroviruses
Exogenization vs Endogenization
by Yingguang Liu and Charles Soper on September 30, 2009Abstract
Scattered in human and animal genomes are a class of repetitious genetic elements called endogenous retroviruses (ERVs), which bear sequence homology to retroviral genomes. They constitute about 8% of the human genome. Evolutionists assume that all endogenous retroviruses are remnants of germ line infection by exogenous retroviruses.
No. "Evolutionists" (meaning scientifically literate people with no reliogio/political axe to grind) conclude, on the basis of copious and diverse evidence, that endogenous retroviruses are the inherited remnants of germ-line DNA integration by exogenous retroviruses. They conclude this on the basis, not of assumptions that arrive out of thin air, nor out of a hatred for any gods, but on the basis of the evidence. See "Why do virologists and geneticists think that ERVs come from retroviruses? Isn't that just supposition on their part?"
However, the essential beneficial function of some ERVs and complex interaction between ERVs and other host DNA sequences suggest that some retroviruses were created in the cell as part of the host genome. The original ERV elements were endowed with the ability to package themselves in proteins and lipids and leave the host cell as infectious particles. Thus we speculate exogenous retroviruses were derived from endogenous retroviruses (exogenization, as opposed to endogenization). The ability of retroviruses to transpose within the host cell or to infect another host may have been designed for protein-coding, for gene regulation, and for DNA repair by homologous and non-homologous recombination. After the Fall, retroviral elements have been degraded by mutations. Retroviral insertion into the host genome also became deregulated, causing insertional mutagenesis. Deregulation of exogenous retroviruses resulted in pathogenesis.
No complete ERVs are known to have beneficial function. The beneficial functions that are known are provided by certain sub-components of ERVs, notably their promoters, and in some cases, their environment genes. Drop a promoter at random into a genome, and there is a good likelihood that it will promote some existing genes. Any such promoters that have a beneficial effect will naturally be selected for.
Speculation is all very well in science. It is the starting point for many scientific topics. But beyond that, speculation without evidence, and especially speculation that cannot be tested, is of no value.
A "Fall" is an integral part of creationist mythology. It cannot be assumed to have happened. Again, speculation without evidence is worthless.
No complete ERVs are known to have beneficial function. The beneficial functions that are known are provided by certain sub-components of ERVs, notably their promoters, and in some cases, their environment genes. Drop a promoter at random into a genome, and there is a good likelihood that it will promote some existing genes. Any such promoters that have a beneficial effect will naturally be selected for.
Speculation is all very well in science. It is the starting point for many scientific topics. But beyond that, speculation without evidence, and especially speculation that cannot be tested, is of no value.
A "Fall" is an integral part of creationist mythology. It cannot be assumed to have happened. Again, speculation without evidence is worthless.
Keywords: endogenous, retroviruses, endogenization, exogenization, history, function, regulation
The Problem of ERVs
Retroviridae is a family of enveloped RNA viruses that utilize reverse transcription during their replication cycle and integrate their DNA genome into host cell chromosomes. The integrated viral DNA (provirus) is characterized by long terminal repeats (LTR) flanking gag,pro, pol, and env genes. Some genera, the complex retroviruses, encode accessory nonstructural proteins such as rev, tat, etc., by alternative splicing.
Endogenous retroviruses (ERVs) are DNA sequences in eukaryotic genomes that are homologous to proviruses in gene sequences and organization. Some are capable of packaging RNA genomes into viral particles for intracellular transposition or intercellular transmission. However, most ERVs, especially human endogenous retroviruses (HERVs), contain premature stop codons, frameshifts, substitutions, and deletions, so that they are incapable of expression, let alone transposition and transmission (Belshaw et al. 2005).
Most biologists assume endogenous retroviruses are remnants of ancestral germ line infections (Belshaw et al. 2005).
Here again is the assertion that it is an assumption. It is not. It is a conclusion based on solid evidence.
Since humans and primates share similar ERVs, not only in sequence but also in position, it is assumed (NO!) that exogenous retroviruses infected the common ancestor of both (Belshaw et al. 2005; Bonnaud et al. 2005). The best alternative explanation is that orthologous ERVs were created to occupy similar genomic loci in separate species by a single designer to carry out similar physiological functions.
Locus specificity is not a requirement for function. Indeed, some retrovirally derived genes that are now essential in some species are different genes from different ERVs in different chromosomes in different lineages! What's that all about? Different designs, different designers? The view of the genome as an intricate interconnected device is deeply mistaken. It is more akin to a box of loose-leaf recipes, some of which have been stolen from viruses.
If ERVs were created in the cell in the beginning, at least some exogenous retroviruses may have been derived from endogenous viruses when viral particles were released from the cell by budding—exogenization. While the ability to exit the host cell and transmit between individuals is designed, uncontrolled infection and deregulated insertion of retroviruses have caused much havoc in the post-Fall world.
Retroviruses integrate their genomes into host DNA in an uncontrolled way, and are therefore more likely to cause serious problems than to do any good. This is why they are not used as gene delivery vectors by known intelligent designers - human gene therapists. There is no evidence that integrase, the retroviral enzyme that performs the integration was ever locus specific, and no evidence that the speculate "Fall" could have the effect of destroying a fictitious and unnecessary "pre-Fall" locus specific integrase. Why would an omnipotent and intelligent designer need to distribute genes in such a roundabout way in any case?
Here again is the assertion that it is an assumption. It is not. It is a conclusion based on solid evidence.
Since humans and primates share similar ERVs, not only in sequence but also in position, it is assumed (NO!) that exogenous retroviruses infected the common ancestor of both (Belshaw et al. 2005; Bonnaud et al. 2005). The best alternative explanation is that orthologous ERVs were created to occupy similar genomic loci in separate species by a single designer to carry out similar physiological functions.
Locus specificity is not a requirement for function. Indeed, some retrovirally derived genes that are now essential in some species are different genes from different ERVs in different chromosomes in different lineages! What's that all about? Different designs, different designers? The view of the genome as an intricate interconnected device is deeply mistaken. It is more akin to a box of loose-leaf recipes, some of which have been stolen from viruses.
If ERVs were created in the cell in the beginning, at least some exogenous retroviruses may have been derived from endogenous viruses when viral particles were released from the cell by budding—exogenization. While the ability to exit the host cell and transmit between individuals is designed, uncontrolled infection and deregulated insertion of retroviruses have caused much havoc in the post-Fall world.
Retroviruses integrate their genomes into host DNA in an uncontrolled way, and are therefore more likely to cause serious problems than to do any good. This is why they are not used as gene delivery vectors by known intelligent designers - human gene therapists. There is no evidence that integrase, the retroviral enzyme that performs the integration was ever locus specific, and no evidence that the speculate "Fall" could have the effect of destroying a fictitious and unnecessary "pre-Fall" locus specific integrase. Why would an omnipotent and intelligent designer need to distribute genes in such a roundabout way in any case?
And God Saw That It Was Good
Beneficial functions of ERVs
Geneticists are increasingly aware of the beneficial functions of transposable elements, including ERVs, for the host cell. ERVs are known to be involved in a wide range of physiological processes of the host. (1) Direct coding of proteins. ERVs are highly expressed in reproductive tissues (ovaries, testes, placenta, etc.) and the early embryo (Arimi et al. 2006; Kjeldbjerg et al. 2008; Peaston et al. 2004). There is a confirmed case of essential ERV function, that is, syncytins, encoded by the env gene of ERVs in human, apes, sheep, and rodents (Mallet et al. 2004; Dunlap et al. 2006; Dupressoir et al. 2005). For exogenous retroviruses, the fusogenic property of the env glycoprotein causes membrane fusion between the viral envelope and the host cell membrane, enabling the viral core to enter the cell. Cells infected by a retrovirus may fuse with one another since env products are expressed on host cell membrane before viral release. The env product of ERVWE1 and ERV-FRD of human and primates, as well as the endogenous Jaagsiekte sheep retrovirus (enJSRV) in sheep and similar ERVs in mice, are expressed on the membrane of trophoblast cells during early embryonic development, leading to cell fusion and formation of syncytiotrophoblast, which is essential for the production of chorionic gonadotropin and development of the placenta (fig. 1) (Frendo et al. 2003; Malassiné et al. 2008). Sheep embryos in which syncytin production is inhibited by antisense oligonucleotides were aborted (Dunlap et al. 2006).
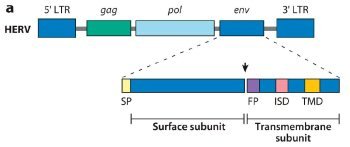
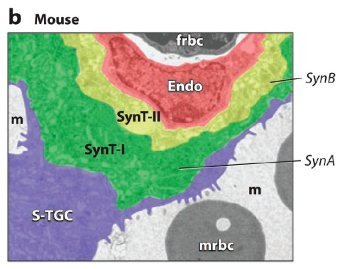
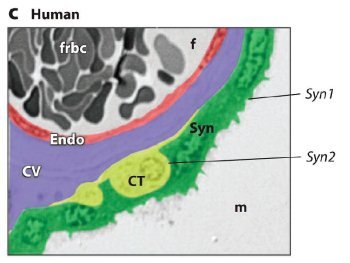
Fig. 1. ERV genetic structure and expression in the murine and human placenta.
(a) Generic structure of a human endogenous retrovirus (HERV), showing the long terminal repeats (LTRs), gag (group-specific antigen gene), pol (polymerase), and env (envelope gene). Syncytins are encoded by the env gene and are composed of surface and transmembrane env subunits. The arrowhead represents a consensus furin cleavage site. SP, signal peptide; FP, fusion peptide; ISD, putative immunosuppressive domain; TMD, transmembrane domain. Schematic of subunits and corresponding domains adapted from Mangeney et al. 2007.
(b) Expression of Syncytin A (SynA) and Syncytin B (SynB) in the murine placenta. Colorized electron micrograph of the mouse placenta showing three layers of fetal trophoblast cells that separate the maternal blood space (m) from the fetal blood space. SynA is restricted to syncytiotrophoblast layer I (SynT-I) and SynB to syncytiotrophoblast layer II (SynT-II). Endo: fetal endothelium; frbc: fetal red blood cell; mrbc: maternal red blood cell; S-TGC: sinusoidal trophoblast giant cell.
(c) Expression of Syncytin 1 (Syn1) and Syncytin 2 (Syn2) in the human placenta. Colorized section of a third-trimester villus depicting fetal cell layers separating the maternal blood space (m) and fetal blood space (f). The human placenta has only a single syncytial layer. Syn1 is predominantly expressed in the syncytiotrophoblast, whereas Syn2 is expressed in the villous cytotrophoblast (CT) cells. CV: chorionic villi; Endo: fetal endothelium; frbc: fetal red blood cells. Reprinted abridged, with kind permission, from the Annual Review of Cell and Developmental Biology, vol. 24. © 2008 by Annual Reviews at www.annualreviews.org
Beside cell fusion, another possible function of ERV proteins is the role of the env glycoprotein in maternal tolerance of the semiallogeneic fetus. Many retroviral env proteins contain a conserved immunosuppressive domain (Good et al. 1999). The human syncytin-2 and the murine syncytin-B have both been shown to be immunosuppressive (Mangeney et al. 2007). In addition, the transmembrane subunit of Molony murine leukemia virus as well as the full-length env protein of HERV-H were able to protect engrafted allogeneic tumor from host immunity in laboratory mice (Mangeney et al. 2001; Mangeney and Heidmann 1998). If as claimed, these sequences were exploited de novo after endogenization of an ERV (exaptation), a burden of proof lies on evolutionists to demonstrate such vital functions could ever arise without catastrophe to the host.
See Abbie Smith's blog entry, "More syncytia sweetness". Primates, mice and sheep each use a different syncytin gene, from the env region of a different ERV, in a different chromosome, in order to build an essential syncytia in the placentas of their respective lineages. (A syncytium is a cell that contains many cell nuclei, as opposed to normal nuclear cells, which usually contain only one cell.) Why different syncytins from different ERVs? Different designs, different designers? Hardly. And why embed the syncytins in ERVS in the first place? What's all the other ERV baggage for?
It turns out that syncytia formation is a native trick of retroviruses. HIV will deal with attacking T-cells by absorbing them into the cells it has already infected. How? By attaching them to itself and joining with them, using the very same env proteins it used to enter the original cell! Clever trick. It avoids all the tedious business of invading every T-cell in order to neutralize them.
Note that the different syncytia belong to different lineages - further proof of evolution. We can even trace their evolution through their respective family trees. See also, Mammals Made by Viruses. Why on earth would a designer take such pains to mimic the the sort of thing we expect from evolution - for no sensible reason? Why did it not mix things up a bit? Why did it never choose to produce any evidence contrary to what we would expect of evolution?
When we look at the human/chimp syncytin gene, and consider it's origin, there are two possibilities.
a) A designer or designers inserted it in the genome of both species, in exactly corresponding locations in the DNA (unnecessarily), the syncytin gene being embedded in an elaborately detailed structure that precisely, and pointlessly mimics a retroviral insertion. (See "Why do virologists and geneticists think that ERVs come from retroviruses?") A designer or designers also inserted a different syncytin, in a different location, in a different chromosome, precisely and pointlessly mimicking a different retroviral insertion in mice. And a completely different one again in sheep. The supposed designer(s) didn't bother with horses. Or pigs. They were designed to get along without a placental syncytiotrophoblast layer, relying instead on a layer consisting of discrete nuclear cells (cells with a single nucleus each).
b) A common ancestor of chimps and humans, its species happily reproducing like horses and like pigs, using epitheliochorial placentas acquired a retroviral infection in a germ-line cell. When it came to reproduce, one of the retroviral genes woke up and affected the development of certain placental cells, having the effect of merging them into a single multi-nucleus cell (a syncytium) just as the corresponding gene in HIV does with T-cells today. This aided reproduction, giving an advantage in the reproductive stakes, such that more and more individuals inherited the trick, and it eventually became a ubiquitous (fixed) feature in the species. At the same time it was spreading through the population, older genes for placental development that were no longer needed mutated into uselessness, rendering the new retroviral gene (but not the rest of its accompanying retroviral baggage) essential. The same thing happened, but with a different retrovirus, in mice. And again, independently, with a different retrovirus in sheep. It did not happen in pigs, or in horses.
Which of the above two scenarios best explains the retroviral-like structure that is to be found at the site of the human/chimp syncytin gene?
It turns out that syncytia formation is a native trick of retroviruses. HIV will deal with attacking T-cells by absorbing them into the cells it has already infected. How? By attaching them to itself and joining with them, using the very same env proteins it used to enter the original cell! Clever trick. It avoids all the tedious business of invading every T-cell in order to neutralize them.
Note that the different syncytia belong to different lineages - further proof of evolution. We can even trace their evolution through their respective family trees. See also, Mammals Made by Viruses. Why on earth would a designer take such pains to mimic the the sort of thing we expect from evolution - for no sensible reason? Why did it not mix things up a bit? Why did it never choose to produce any evidence contrary to what we would expect of evolution?
When we look at the human/chimp syncytin gene, and consider it's origin, there are two possibilities.
a) A designer or designers inserted it in the genome of both species, in exactly corresponding locations in the DNA (unnecessarily), the syncytin gene being embedded in an elaborately detailed structure that precisely, and pointlessly mimics a retroviral insertion. (See "Why do virologists and geneticists think that ERVs come from retroviruses?") A designer or designers also inserted a different syncytin, in a different location, in a different chromosome, precisely and pointlessly mimicking a different retroviral insertion in mice. And a completely different one again in sheep. The supposed designer(s) didn't bother with horses. Or pigs. They were designed to get along without a placental syncytiotrophoblast layer, relying instead on a layer consisting of discrete nuclear cells (cells with a single nucleus each).
b) A common ancestor of chimps and humans, its species happily reproducing like horses and like pigs, using epitheliochorial placentas acquired a retroviral infection in a germ-line cell. When it came to reproduce, one of the retroviral genes woke up and affected the development of certain placental cells, having the effect of merging them into a single multi-nucleus cell (a syncytium) just as the corresponding gene in HIV does with T-cells today. This aided reproduction, giving an advantage in the reproductive stakes, such that more and more individuals inherited the trick, and it eventually became a ubiquitous (fixed) feature in the species. At the same time it was spreading through the population, older genes for placental development that were no longer needed mutated into uselessness, rendering the new retroviral gene (but not the rest of its accompanying retroviral baggage) essential. The same thing happened, but with a different retrovirus, in mice. And again, independently, with a different retrovirus in sheep. It did not happen in pigs, or in horses.
Which of the above two scenarios best explains the retroviral-like structure that is to be found at the site of the human/chimp syncytin gene?
(2) Gene regulation. ERVs, especially their LTR, may provide regulatory sequences for nearby genes (Conley, Piriyapongsa, and Jordan 2008). Multiplicity of ERV copies offers synchronized regulation of interspersed genes (regulatory suites) (Shapiro 2005); while transposition of ERVs confers flexibility to the genome, reshaping it at different stages of life and in different environments (Borger 2009a). The former is exemplified by the finding that ERVs offer alternative promoters and first exons for a significant proportion of genes expressed in the oocyte and early embryonic cells of the mouse (Peaston 2004), while the latter is seen in yeast Ty elements, which are more actively transposing than animal ERVs.
The yeast Ty elements are homologous to animal ERVs, except they do not have an env gene and therefore are never transmitted between cells. Like many ERVs, the Ty1 elements are activated during sexual reproduction. Most Ty elements are repressed in diploid cells. In haploid cells, Ty3 and Ty5 are mobilized by pheromones through multiple pheromone-response elements in their 5' LTR (Lesage and Todeschini 2005). Since the yeast does not form complex reproductive structures such as the placenta, the role of Ty elements in sexual reproduction should be either for gene regulation or for DNA recombination (see below on DNA repair and recombination). In addition, the Ty1 elements are mobilized under nutrient depletion to induce invasive/filamentous growth. Interestingly, Ty1 contains a filamentous responsive element (FRE) superimposed on the coding sequence of a structural gene (TY1A, a homolog of gag), and the FRE responds to specific transcription factors of the cell to activate cellular genes downstream of the insertion site. Because the FRE sequence is multifunctional (both coding and regulatory), it is multiply constrained, which undermines the proposal that FRE may have evolved within a proviral gene.
The pol polyprotein of ERVs, which includes the reverse transcriptase, integrase, etc., is essential for retrotransposition. In the case of yeast Ty elements, the viral capsid proteins (homologues of gag products) are also essential for mobilization, as they form the virus-like particle (VLP), in which reverse transcription occurs. The capsid proteins may also guide and protect the cDNA before integration (Lesage and Todeschini 2005).
LTRs are promoters. The interesting thing about them is that, unlike "native" promoters, that can be transcribed back again into RNA. Why is this interesting? Because it is essential for the retroviral replication cycle. A provirus integrated into the host DNA will not be transcribed without them. Now if you drop a promoter at random into the genome, (integrase is incapable of targeting specific DNA loci) it is quite likely to promote native genes too. This provides a source of variation for natural selection to work on. This is all that needs to be understood when we look at LTRs performing adaptive regulatory functions.
(3) DNA repair and recombination. ERV activities are often associated with DNA damages and play a role in repair mechanisms. It has been shown that ERVs are activated when the cell is irradiated with UV or treated with chemical mutagens (Hohenadl et al. 1999; Hsieh and Weinstein 1990; Lesage and Todeschini 2005). One piece of evidence that ERVs are involved in DNA repair is that ERV fragments, including entire LTRs, are sometimes captured between reconnected ends of double-strand breaks (Lesage and Todeschini 2005; Lin and Waldman 2001). (This indicates that not all solo LTRs are results of ERV deletions). Since double-strand breaks are involved in important cellular functions such as meiotic recombination, the availability of ERVs and other repetitious elements in certain chromosomal regions may constitute the structural basis of recombinational hotspots (Lesage and Todeschini 2005). In addition, newly synthesized cDNA of the yeast Ty elements are known to recombine with pre-existing elements based on sequence homology. This process can effectively repair degenerated ERV elements. The same mechanism of recombinational repair of ERVs may also exist in human and animals. The env gene is the best preserved structural gene in many human and animals ERVs. Enabling intercellular transmission of retroviruses, the env gene provides a mechanism for cells to repair ERV genes using an exogenous virus. Without the env gene (for example, intracisternal A-type particles of mouse whoseenv gene has been degraded), ERVs can only repair each other within the same cell by RNA-mediated gene conversion.
Obviously, recombination between proviral elements at non-homologous chromosomal positions (ectopic recombination) will create large-scale chromosomal rearrangements. This is rare, presumably prevented by separation of chromosomal domains within the nucleus and recombination control mechanisms.
Is there some point to all this?
Is there some point to all this?
(4) Transduction. Viruses may occasionally package genetic fragments of the host cell into progeny viral particles and carry them into a new host or to new loci within the same host genome, a process known as transduction. The avian leukosis virus (ALV) can transduce small RNA genes between hosts (Hajjar and Linial 1993). Some members of HERV-K14I are found to carry portions of a cellular gene called transient receptor potential channel 6 (TRPC6), and is probably responsible for the spreading of the sequences in the human genome (Flockerzi et al. 2005). While viral transduction is an important mechanism of horizontal gene transfer in bacteria, the biological significance of retroviral transduction in eukaryotes is unclear.
OR this?
(5) Resistance to exogenous retroviruses. If an ERV is expressed in a cell, re-infection by a related exogenous retrovirus is prevented through interference. The env protein of an ERV that is inserted into the cell membrane will interfere with the corresponding exogenous virus by receptor competition. This protects the cell from being overloaded with retroviruses. For example, enJSRVs can block the entry of exogenous JSRVs because they all utilize the cellular hyaluronidase-2 as a receptor (Spencer et al. 2003). It is noteworthy that defective ERVs are no less interfering. Two enJSRVs, enJS56A1 and enJSRV-20, contain a mutant gag polyprotein that can interfere with the late stage replication of exogenous JSRVs (Arnaud et al. 2007). Thus interference between defective and replication-competent retroviruses provides an important mechanism of ERV copy number control.
So retroviruses were designed to protect us from - retroviruses! I've heard of intelligent design and dumb design, but futile and pointless design?
(5) Resistance to exogenous retroviruses. If an ERV is expressed in a cell, re-infection by a related exogenous retrovirus is prevented through interference. The env protein of an ERV that is inserted into the cell membrane will interfere with the corresponding exogenous virus by receptor competition. This protects the cell from being overloaded with retroviruses. For example, enJSRVs can block the entry of exogenous JSRVs because they all utilize the cellular hyaluronidase-2 as a receptor (Spencer et al. 2003). It is noteworthy that defective ERVs are no less interfering. Two enJSRVs, enJS56A1 and enJSRV-20, contain a mutant gag polyprotein that can interfere with the late stage replication of exogenous JSRVs (Arnaud et al. 2007). Thus interference between defective and replication-competent retroviruses provides an important mechanism of ERV copy number control.
So retroviruses were designed to protect us from - retroviruses! I've heard of intelligent design and dumb design, but futile and pointless design?
Regulation of ERVs by the cell
Complex collaborations between ERVs and other components of the eukaryotic genome further argue for the creation of ERVs as parts of the cell. In the case of human syncytin-1, sequences from another ERV, MaLR, provides a placenta-specific enhancer, which, in collaboration with the LTR of ERVWE1 itself, regulates the temporal and spatial expression of the syncytin (Bonnaud et al. 2005; Prudhomme, Oriol, and Mallet 2004). While the regulatory sequences are targets of general and placenta-specific transcription factors, syncytin proteins must recognize the right membrane receptor to induce fusion of cytotrophoblasts (Esnault et al. 2008; Yu et al. 2002). All of these viral and non-viral factors must be present simultaneously for placenta development.
An env gene from one ERV, promoted by an LTR from another ERV doesn't look like design. What's the rest of the retroviral baggage for? And as has been pointed out above, why design different syncytins from different ERVs in different chromosomes in different lineages? Common design, common designer? Hardly!
An env gene from one ERV, promoted by an LTR from another ERV doesn't look like design. What's the rest of the retroviral baggage for? And as has been pointed out above, why design different syncytins from different ERVs in different chromosomes in different lineages? Common design, common designer? Hardly!
Transposition of yeast Ty elements occurs at a fairly low frequency (10-7–10-8 per generation) (Lewin 2004), and is activated by cellular and environmental signals such as DNA damage, nutrient depletion, and pheromones (Lesage and Todeschini 2005). Transposition of ERVs in modern human genomes is rarely documented, if it occurs at all. Activities of human and animal ERVs are primarily regulated at the transcription level. Just as the LTR of some yeast Ty elements contains pheromone responsive elements, the LTR of some HERVs contains putative steroid response elements (Ono, Kawakami, and Ushikubo 1987). Transcription of HERVs responds to sex hormones, demonstrating a temporal pattern during the menstrual cycle of a female (Loggans, Mudge, and Liu 2009; Ono, Kawakami, and Ushikubo 1987). Epigenetic control also plays a key role in the regulation of ERVs (Schulz, Seinhoff, and Flori 2006). Global demethylation and remethylation of the cellular genome during gametogenesis and early embryotic development leave a window of ERV de-repression. Transposition during gametogenesis is less detrimental to the host species, because of the overwhelming surplus of gametes, especially sperms.
It has been noticed recently that integration of human and animal retroviruses into the host genome is not entirely random. For example, the murine leukemia virus prefers transcription start sites, while the human immunodeficiency virus prefers to insert in actively transcribed genes (Mitchell et al. 2004). While the preferred sites of modern pathogenic retroviruses may have deviated from the intended targets at the time of their creation, the phenomenon does suggest precise insertion as a designed feature of retroviruses. The yeast Ty elements demonstrate even stricter target site preference. Some Ty elements reside upstream of genes transcribed by the eukaryotic RNA polymerase III (Pol III), such as tRNA genes; others are found in heterochromatin regions such as telomeres (Lesage and Todeschini 2005). Targeting of Ty3 requires interaction between Pol III transcription factors (TFIIIB and TFIIIC) and the viral integrase, while insertion of Ty5 involves interaction between a chromatin structural protein (Sir4) and the integrase. Even the orientation of Ty insertion is specified by host proteins (Rinckel and Garfinkel 1996). Insertion within heterochromatin represses transcription of the Ty elements, which runs contrary to the conception that retroposons are “selfish” elements whose only goal is to increase their own copy numbers at the cost of the host cell. Directed integration may also partially explain the positional “conservation” of orthologous ERVs in human and primate genomes.
Another aspect of ERV regulation is copy number control. There is a genome-stabilization mechanism known as homology-dependent gene silencing (also called cosuppression) that inhibits the mobility of transposable elements as their copy numbers increase (Reiss and Mager 2007). As mentioned above, expressed ERVs can interfere with re-infection of corresponding exogenous retroviruses thereby preventing further endogenization. Ty elements exhibit strong cosuppression even without transcription of the suppressing element, through a mechanism that may involve direct Ty-Ty interactions (Garfinkel et al. 2003).
A Tale of Two ERV “Cousins”
The findings of comparative genomics around the syncytin-1 loci is especially challenging to creationists. The gene is well conserved among hominoids— humans, chimpanzees, gorillas, orangutans, and gibbons, but not in the Old World Monkeys (OWMs, also known as Cercopithecidae, such as baboons and rhesus monkeys) or New World Monkeys (NWMs, also known as Platyrrhines, such as marmosets and galagoes) (Bonnaud et al. 2005; Caceres 2006; Mallet et al. 2004). While it is conceivable that monkeys use mechanisms of placenta development that are different from those of the hominoids, what is most puzzling is that OWMs do have an orthologous ERV-W element but it has been degraded by nonsense mutations and frameshifts, and therefore not encoding an active syncytin protein. So it appears that an ERV-W provirus “integrated into the germ line of a Catarrhine ancestor,” that is, a common ancestor of Cercopitheques and hominoids, followed by “two different evolutionary pathways in Cercopithecidae and hominoids, a genetic drift versus domestication, respectively” (Bonnaud et al. 2005). So the presence of an inactivated ERV-W in OWMs simultaneously argues for the idea that ERVs are optional add-ins to the primate genome and the idea that humans and monkeys have a common Catarrhine ancestor!
But is the monkey ERV-W really nonfunctional? A study of up-regulated genes in the endometrium of female rhesus monkeys during their monthly window of receptivity identified a transcript that is 92% identical to the human syncytin-1 (Okulicz and Ace 2003). The temporal pattern of its expression indicates a function of the element in simian reproduction, even though it cannot be translated into a complete protein. Many genes in the genome are transcribed but not translated (Sanford 2008). Apparently degraded open reading frames do not necessarily mean junk sequences. Most DNA sequences in the genome are multifunctional. Besides encoding RNA or proteins, they must simultaneously form genome-wide patterns such as nucleosome binding sites, which is probably another reason that ERVs must be repetitious.
So the ERV-W elements in OWMs and hominoids are not necessarily related by descent, but are created to perform related functions in different primate families. This view is supported by another related finding of an ERV-H element immediately upstream of the ERV-W element. Unlike the ERV-W, the ERV-H element is present in NWMs, as well as in Catarrhines. However, the element is so different between Platyrrhines and Catarrhines that different symbols, ERV-H(p) and ERV-H(c), are used, and they are considered “lineage specific,” implying independent origins (Bonnaud et al. 2005). So even evolutionists sometimes acknowledge that orthologous sequences at similar loci are not necessarily phylogenetic twins.
Monkeys’ placentas require syncytiotrophoblasts. There is another syncytin gene, syncytin-2, encoded by the env protein of ERV-FRD, which is conserved in human, apes, OWMs, and NWMs (Blaise et al. 2003). Syncytin-2 has immunosuppressive as well as fusogenic properties, suggesting its involvement in maternal tolerance of the fetus; while syncytin-1 is only fusogenic (Mangeney et al. 2007). Like many other physiological processes, cell fusion during placenta development is designed to be accomplished by redundant genes. In addition to syncytin-2, monkeys may have other syncytins encoded by genes in different loci. Interestingly, mice also have two syncytins (syncytin-A and B), encoded by ERVs that are not orthologous to ERVWE1 or ERV-FRD (Dupressoir et al. 2005). One of them (syncytin-B) is immunosuppressive, and the other (syncytin-A) is not (Mangeney et al. 2007). Syncytin is encoded by yet another unrelated ERV, enJSRV, in sheep. Because the lack of syncytin homology does not support common ancestry of rodents, ruminants, and primates, evolutionists tautologously describe this as convergent evolution. It is better understood as a common placentation blueprint employing multiple ERVs in multiple mammalian orders.
“Spontaneous Generation” of Retroviruses
If ERVs were indeed created in the beginning, were modern exogenous retroviruses derived from ERVs? The concept of exogenization is not new. Prions have been thought to arise from mutation of a normal cellular gene, and the mutated form can then be transmitted horizontally to another host (Wadsworth et al. 2003). Early in the quest for endogenous retroviruses, it was discovered that treating normal chicken cells with ionizing radiation or carcinogens stimulated the release of infectious ALV (Weiss et al. 1971). Likewise, the murine leukemia virus (MLV) can be induced from virus-free cell cultures chemically (Lowy et al. 1971). The switch between horizontal and vertical (genetic) transmission of retroviruses present a chicken-egg enigma of origins. Since ERVs are shown to be integral functional parts of cellular genomes, endogenous viruses predate exogenous ones, although simultaneous creation of endogenous and extracellular viruses cannot be ruled out.
The biggest challenge to the idea of exogenization is that exogenous retroviruses include groups of complex retroviruses which have no endogenous counterparts. The fifth edition of Field’s virology lists seven retroviral genera, namely, Alpharetrovirus, Betaretrovirus, Gammaretrovirus, Deltaretrovirus, Epsilonretrovirus, Lentivirus, and Spumavirus(Goff 2001). The first three genera are considered simple retroviruses, while the rest are considered complex. Complex retroviruses have small accessory proteins encoded by alternatively spliced transcripts that are absent in simple retroviruses, such as rex and tax of Deltaretrovirus, tat, rev, vpr, vpu, vif and nef of Lentivirus, tas/bel-1 and bet ofSpumavirus. These accessory proteins are nonstructural proteins involved in viral replication or pathogenesis.
There are three classes of HERVs corresponding to Betaretrovirus, Gammaretrovirus and Spumavirus, most of which are simple viruses. HERV-L, which is related to Spumavirus, does not have a detectable env gene, let alone accessory genes which are typically encoded by alternatively spliced env transcripts (Cordonnier, Casella, and Heidmann 1995). No HERVs are found in the other retroviral genera, even though these genera include exogenous human viruses. For example, there are no endogenous human lentiviruses, even though HIV is the best known exogenous human retrovirus. How might the exogenization hypothesis explain the origin of some complex retroviruses with their accessory proteins?
First, some HERVs do encode accessory proteins. Even though HERV-Ks are homologous to Betaretrovirus, they encode an accessory protein called rec, whose function is analogous to rex of Deltaretrovirus and rev of Lentivirus, transporting unspliced and singly spliced viral transcripts out of the nucleus for translation (Magin-Lackmann et al 2001). Rec may not be the direct ancestor of rex or rev, but it proves that ERVs are not necessarily simple, and they may contain, or did contain, ancestral accessory genes. Second, there are complex ERVs in animals. Recently, an endogenous lentivirus was discovered in European rabbits (Katzourakis et al. 2007). The element contains tat and revin locations similar to those of exogenous lentiviruses. Most other features of Lentivirus are also well preserved, including the rev responsive element. Katzourakis et al stated,
Based upon the above, it is tempting to speculate that lentiviruses in rabbits, or perhaps other, related lagomorphs, might represent the precursors of modern exogenous lentiviruses. In accordance with this idea, the ancestral geographic range of the European rabbit (southern Europe and northwest Africa) overlaps that of many species now harboring exogenous lentiviruses, including cattle, horses, and (wild) cats (Katzourakis et al. 2007).
So the hypothesis of exogenization depends on cross-species infection of retroviruses. This is not a new idea either, since HIV has long been speculated to be derived from simian immunodeficiency viruses. There are numerous cases of non-parallel relationships between the phylogeny of the ERVs and the phylogeny of the host species. In a study of ERV-W LTRs in hominoids (Huh et al. 2003), the LTRs did not cluster according to host species, but demonstrated a wide overlap across species. One LTR element was 100% identical between orangutan and gibbon sequences. The authors of the report explained these findings as evidences of “independent evolution” between host and viruses. Moreover, there are indications of ERVs jumping across host species over large taxa spans. For instance, the baboon ERV, BaEV, is very closely related to the endogenous RD114 virus in cats by antigenicity and sequence homology. Likewise, the koala endogenous retrovirus, KoRV, is highly similar to the gibbon endogenous retrovirus, GaRV (Weiss 2006). There is even a type of ERVs, known as xenotropic viruses, which do not readily re-infect cells of their own host, but prefer cells of other species in vitro and in vivo. For example, the endogenous ALV of chickens prefers cells of quail, pheasants, and turkey to chicken cells. Xenotrophic ERVs may have been created to exchange genetic elements between species, but their ecological significance remains unclear.
“Thou shalt surely die.”
Degeneration
In his book Genetic entropy and the mystery of the genome, Dr. John Sanford pointed out the degeneration of genomes and predicted the extinction of the human race (Sanford 2008). So the apparent degeneration of ERV elements in vertebrate genomes is real. All known HERVs show varying degrees of deletions and substitutions. Oftentimes, the original viral genes are mutated beyond recognition (Cordonnier, Casella, and Heidmann 1995). Analysis of non-synonymous and synonymous mutations showed that HERV elements have been subjected to purifying selection (Belshaw et al. 2004). Selection may be on the level of the virus, for the propagation of the fittest virus, or on the level of the host, for the preservation of the fittest host. For those ERVs whose genes are only partially preserved, such as ERVWE1, selection seems to be based on their benefit to the host. Homologous recombination between degraded ERV genes and nascent cDNA of replication-competent retroviruses may have slowed down the degeneration of ERVs. Since selection on replicating viruses is more affordable, simultaneous existence of exogenous retroviruses provides a means to preserve ERV genes.
Global degeneration of ERVs is attested to by efforts to regenerate HERV-K(HML-2) (Lee and Bieniasz 2007). Lee and Bieniasz first attempted to generate an infectious HERV-K113, an element that has intact open reading frames (ORFs). However, as they tried to express the viral structural proteins with plasmid constructs, the proteins were poorly expressed and inefficiently processed, leading them to conclude that intact ORFs do not mean intact functions. Later they took a different approach, based on the assumption that all ERV elements have sustained random mutations. By aligning ten relatively intact HERV-K(HML2) elements, including HERV-K113, they generated a consensus sequence and artificially synthesized a presumed “ancestral ERV” (HERVK CON), which proved to be infectious! Viral particles were released from the packaging cell line, and were able to form loci in target cells. Therefore, it appears that at least some HERV elements were created to be infectious.
Degeneration of host and viral genomes has led to deregulation of ERVs. For example, some vitamin-synthesizing genes in the human genome have degenerated, leading to our dependence on dietary vitamins (Borger 2009b). There is a report that modification of histones in human and mouse cells by a vitamin (biotin) suppresses the transcription of ERV elements, and deficiency of the vitamin enhances ERV activities in human and Drosophila, destabilizing chromosomes in cell cultures (Chew et al. 2008). As to Ty elements, mutation of diverse yeast genes that affect chromatin structure/function de-represses their transposition (Nyswaner et al. 2008). In addition to enhanced activities, imprecise, random, or even perverted ERV insertions in the post-Fall world would be expected to cause devastating mutations, which we now briefly consider.
Pathogenesis
Retroviral activities after the Fall disrupt cellular functions. From the time they were discovered, retroviruses have been known to carry oncogenes which directly cause cancer in the infected host (Goff 2001). Endogenous retroviruses in modern genomes are generally oriented away from genes, on the complementary strand of DNA antisense to genes, or in introns (van de Lagemaat, Medstrand, and Mager 2006). This may be due to design rather than selection, and it implies the detrimental nature of random insertions. There might have been perfectly controlled retrotransposition in the beginning, but afterwards deregulated retroviral insertions have been linked to mutations and carcinogenesis. In one report, the 3' LTR of a retroviral gene inserted in the c-Ha-ras proto-oncogene of rat caused over-expression of the gene and was responsible for development of mammary cancer (Bera et al. 1998). In this case, the carcinogen, MNU, worked through activation of the endogenous retroviral element. Moreover, it has been proven in transgenic mice that retroviral insertion can cause recessive lethal mutations (Spence et al. 1989). In humans, HERV-K was once called human teratocarcinoma-derived virus (HTDV) because viral particles were first observed in human teratocarcinoma cell lines (Bannert and Kurth 2004). There are other evidences of HERV involvement in human cancer, although the causal relationship between retroviral activation and carcinogenesis is still foggy (see Nelson et al. 2003 for review). HERV activity has also been connected to autoimmune diseases (Nelson et al. 2003).
Re-endogenization
Why are murine and avian ERVs more intact than HERVs? Infectious ALV and MLV can be activated from normal animal cells by mere radiation or chemical treatment, while it took great efforts to generate an infectious HERV. Are animal ERVs really younger? At least some animal ERVs should be just as old as HERVs, such as those that encode syncytin-A and B of Muridae. Indeed, the ERVs containing syncytin-A and B are both of single copy and “extremely degenerate,” without recognizable gag, with short pol stretches and solitary LTRs (Dupressoir et al. 2005). Despite that the mouse genomes is littered with abundant ERVs, few are currently known to be beneficial. So it is possible that some of the ERV categories, especially the more intact ones, are results of recent invasions. It has been demonstrated that MLVs that infect the ovary or embryo can be endogenized and transmitted as Mendelian genes (Jaenisch 1976; Lock et al. 1988). Many HERV loci exhibit allelic frequency polymorphism and positional polymorphism, which may reflect recent proliferation of ERV elements, whether by re-infection of germ line or retrotransposition (Hughes and Coffin 2004; Turner et al. 2001). Re-endogenization of exogenized retroviruses may not have been harmful before the Fall if proviral insertion was precisely controlled, but genomic insertion of modern exogenous retroviruses, especially deregulated insertion of fallen viruses from a different species against which there is no immunity, should now be mostly destructive. Evolutionists coined the words “molecular domestication” to account for the beneficial function of ERVs after supposedly random endogenization. While this cannot be totally ruled out, it is an exaggeration of its creative power to say “humans are descended from viruses as well as from apes” (Weiss 2006).
Conclusion
ERVs were created to encode co-regulated proteins and to regulate dispersed host genes. Retrotransposition adds to the flexibility of the cellular genome, while intercellular transmission enables ERVs in horizontal gene transfer and homologous repair. Common design and controlled activities may explain the similarities between human and primate ERVs, while deregulation in viral replication and integration is responsible for the pathogenesis of modern retroviruses. Re-endogenization of degenerated exogenous retroviruses is mostly detrimental.
Predictions and Expectations
(1) Despite massive degeneration since the Fall, creationists expect to find more examples of complex, interdependent functions between ERVs and the host genome, which challenges the conception that ERVs are add-ins to pre-existing genomes.
(2) Discoveries are expected concerning the details of the interaction between ERVs and host cell DNA repair and maintenance, which would not be anticipated if ERVs were originally “selfish” exogenous entities.
(3) We expect more examples of degenerate and impaired functions, which may be repaired or restored by relatively small modifications (as with HERV-KCON (Lee and Bieniasz 2007).
(4) In line with their original design, creationists anticipate more examples of functional gene transfer by retroviruses between cells of the same host, between members of the same species, and possibly even between different species.
References
Arimi, M. M., A. Nyachieo, D. K. Langat, A. M. Abdi, and J. M. Mwenda. 2006. Evidence for expression of endogenous retroviral sequences on primate reproductive tissues and detection of cross-reactive ERVs antigens in the baboon ovary: a review. East African Medical Journal 83, no. 2:106–112.
Arnaud, F., M. Caporale, M. Varela, R. Biek, B. Chessa, A. Alberti, M. Golder, M. Mura, Y. Zhang, L. Yu, F. Pereira, J. C. DeMartini, K. Leymaster, T. E. Spencer, and M. Palmarini. 2007. A paradigm for virus–host coevolution: Sequential counter-adaptations between endogenous and exogenous retroviruses. PLoS Pathogens 3: e170.
Bannert, N. and R. Kurth, 2004. Retroelements and the human genome: New perspectives on an old relation.Proceedings of the National Academy of Sciences, USA 101:14572–14579.
Belshaw, R., A. L. Dawson, J. Woolven-Allen, J. Redding, A. Burt, and M. Tristem. 2005. Genomewide screening reveals high levels of insertional polymorphism in the human endogenous retrovirus family HERV-K(HML2): implications for present-day activity. Journal of Virology 79:12507–12514.
Belshaw, R., V. Pereira, A. Katzourakis, G. Talbot, J. Paces, A. Burt, and M. Tristem. 2004. Long term reinfection of the human genome by endogenous retroviruses. Proceedings of the National Academy of Sciences, USA 101:4894–4899.
Bera, T. K., T. Tsukamoto, D. K. Panda, T. Huang, R. C. Guzman, S.-I. Hwang, and S. Nandi. 1998. Defective retroviral insertion activates c-Ha-ras proto-oncogene in an MNU-induced rat mammary carcinoma. Biochemical and Biophysical Research Communications 248:835–840.
Blaise, S., N. de Parseval, L. Benit, and T. Heidmann. 2003. Genomewide screening for fusogenic human endogenous retrovirus envelopes identifies syncytin 2, a gene conserved on primate evolution. Proceedings of the National Academy of Sciences, USA 100:13013–13018.
Bonnaud, B., J. Beliaeff, O. Bouton, G. Oriol, L. Duret, and F. Mallet. 2005. Natural history of the ERVWE1 endogenous retroviral locus. Retrovirology 2:57.
Borger, P. 2009a. The design of life: Part 3—an introduction to variation-inducing genetic elements. Journal of Creation23:99–106.
Borger, P. 2009b. The design of life: Part 4—variation-inducing genetic elements and their functions. Journal of Creation 23:107–114.
Caceres, M. 2006. NISC comparative sequencing program, and J. W. Thomas. 2006. The gene of retroviral origin syncytin 1 is specific to hominoids and is inactive in old world monkeys. Journal of Heredity 97:100–106.
Chew, Y. C., J. T. West, S. J. Kratzer, A. M. Ilvarsonn, J. C. Eissenberg, B. J. Dave, D. Klinkebiel, J. K. Christman, and J. Zemnleni. 2008. Biotinylation of histones represses transposable elements in human and mouse cells and cell lines and in Drosophila melanogaster. The Journal of Nutrition 138:2316–2322.
Conley, A. B., J. Piriyapongsa, and I. K. Jordan. 2008. Retroviral promotors in the human genome. Bioinformatics24:1563–1567.
Cordonnier, A., J.-F. Casella, and T. Heidmann. 1995. Isolation of novel human endogenous retrovirus-like elements with foamy virus-related pol sequence. Journal of Virology 69:5890–5897.
Dunlap, K. A., M. Palmarini, M. Varela, R. C. Burghardt, K. Hayashi, J. K. Farmer, and T. E. Spencer. 2006. Endogenous retroviruses regulate periimplantation placental growth and differentiation. Proceedings of the National Academy of Sciences, USA 103:14390–14395.
Dupressoir, A., G. Marceau, C. Vernochet, L. Benit, C. Kanellopoulos, V. Sapin, and T. Heidmann. 2005. Syncytin- A and syncytin-B, two fusogenic placenta-specific murine envelope genes of retroviral origin conserved in Muridae,Proceedings of the National Academy of Sciences, USA 102:725–730.
Esnault, C., S. Priet, D. Ribet, C. Vernochet, T. Bruls, C. Lavialle, J. Weissenbach, and T. Heidmann, T. A. 2008. Placenta-specific receptor for the fusogenic, endogenous retrovirus-derived, human syncytin-2. Proceedings of the National Academy of Sciences, USA 105:17532–17537.
Flockerzi, A., S. Burkhardt, W. Schempp, E. Meese, and J. Mayer. 2005. Human endogenous retrovirus HERV-K14 families: status, variants, evolution, and mobilization of other cellular sequences. Journal of Virology 79: 2941–2949.
Frendo, J. L., D. Olivier, V. Cheynet, J. L. Blond, O. Bouton, M. Vidaud, M. Rabreau, D. Evain-Brion, and F. Mallet. 2003. Direct involvement of HERV-W Env glycoprotein in human trophoblast cell fusion and differentiation. Molecular and Cellular Biology 23:3566–3574.
Garfinkel, D. J., K. Nyswaner, J. Wang, and J. Y. Cho. 2003. Posttranscriptional cosuppression of Ty1 retrotransposition. Genetics 165:83–99.
Goff, S. P. 2001. Retroviridae: The retroviruses and their replication. In Fields virology, eds. D. M. Knipe, P. M. Howley, D. E. Griffin, M. A. Martin, R. A. Lamb, B. Roizman, and S. E. Straus, 5th ed., pp. 1999–2069. Philadelphia, Pennsylvania: Lippincott Williams and Wilkins.
Good, R. A., M. Ogasawara, W. T. Liu, E. Lorenz, and N. K. Day. 1990. Immunosuppressive actions of retroviruses.Lymphology 23:56–59.
Hajjar, A. M., and M. L. Linial. 1993. Characterization of a unique retroviral recombinant containing 7S L sequences.Journal of Virology 67:7677–7679.
Hohenadl, C., H. Germaier, M. Walchner, M. Hagenhofer, M. Herrmann, M. Stürzl, P. Kind, R. Hehlmann, V. Erfle, and C. Leib-Mösch. 1999. Transcriptional activation of endogenous retroviral sequences in human epidermal keratinocytes by UVB irradiation. The Journal of Investigative Dermatology 113:587–594.
Hsieh, L. L. and I. B. Weinstein. 1990. Factors influencing the expression of endogenous retrovirus-like sequences in Rat 6 cells. Molecular Carcinogenesis 3:344–349.
Hughes, J. F. and J. M. Coffin. 2004. Human endogenous retrovirus K solo-LTR formation and insertional polymorphism: implications for human and viral evolution, Proceedings of the National Academy of Sciences, USA101:1668–1672.
Huh, J. W., K. W. Hong, J. M. Yi, T. H. Kim, O. Takenaka, W. H. Lee, and H. S. Kim. 2003. Molecular phylogeny and evolution of the human endogenous retrovirus HERV-W LTR family in hominoid primates. Molecular Cells 15: 122–126.
Jaenisch, R. F. 1976. Germline integration and Mendelian transmission of the exogenous Moloney leukemia virus.Proceedings of the National Academy of Sciences, USA 73: 1260–1264.
Katzourakis, A., M. Tristem, O. G. Pybus, and R. J. Gifford. 2007. Discovery and analysis of the first endogenous lentivirus. Proceedings of the National Academy of Sciences, USA 104:6261–6265.
Kjeldbjerg, A. L., P. Villesen, L. Aagaard, and F. S. Pedersen. 2008. Gene conversion and purifying selection of a placenta-specific ERV-V envelope gene during simian evolution. BMC Evolutionary Biology 8:266.
Lee, N. and P. D. Bieniasz. 2007. Reconstitution of an infectious human endogenous retrovirus. PLoS Pathogens 3: 0119–0130.
Lesage, P. and A. L. Todeschini. 2005. Happy together: The life and times of Ty retrotransposons and their hosts.Cytogenetic and Genome Research 110:70–90.
Lewin, B. 2004. Genes VIII (p. 501). Upper Saddle River, New Jersey: Pearson Education.
Lin, Y. and A. S. Waldman. 2001. Capture of DNA sequences at double-strand breaks in mammalian chromosomes.Genetics 158:1665–1674.
Lock, L. F., E. Keshet, D. J. Gilbert, N. A. Jenkins, and N. G. Copeland. 1988. Studies of the mechanisms of spontaneous germline ecotropic provirus acquisition in mice. The EMBO Journal 7:4169–4177.
Loggans, L., A. Mudge, and Y. Liu. 2009. Expression of human endogenous retroviruses during the menstrual cycle.Occasional Papers of the BSG 13:5–6.
Lowy, D. R., W. P. Row, N. Teich, and J. W. Hartley. 1971. Murine leukemia virus: high-frequency activation in vitro by 5-iododeoxyuridine and 5-bromodeoxyuridine. Science 174:155–156.
Magin-Lackmann, C., S. Hahn, H. Strobel, U. Held, J. Lower, and R. Lower. 2001. Rec (formerly Corf) function requires interaction with a complex, folded RNA structure within its responsive element rather than binding to a discrete specific binding site. Journal of Virology 75:10359–10371.
Malassiné, A., J. L. Frendo, S. Blaise, K. Handschuh, P. Gerbaud, V. Tsatsaris, T. Heidmann, and D. Evain-Brion. 2008. Human endogenous retrovirus-FRD envelope protein (syncytin 2) expression in normal and trisomy 21-affected placenta. Retrovirology 5:6.
Mallet, F., B. Oliver, S. Prudhomme, V. Cheynet, G. Oriol, B. Bonnaud, G. Lucotte, L. Duret, and B. Mandrand. 2004. The endogenous retroviral locus ERVWE1 is a bona fide gene involved in hominoid placental physiology. Proceedings of the National Academy of Sciences, USA 101:1731–1736.
Mangeney, M., N. de Parseval, G. Thomas, and T. Heidmann. 2001. The full-length envelope of an HERV-H human endogenous retrovirus has immunosuppressive properties. Journal of General Virology 82:2515–2518.
Mangeney, M. and T. Heidmann. 1998. Tumor cells expressing a retroviral envelope escape immune rejection in vivo.Proceedings of National Academy of Sciences, USA 95:14920–14925.
Mangeney, M., M. Renard, G. Schlecht-Louf, I. Bouallaga, O. Heidmann, C. Letzelter, A. Richaud, B. Ducos, and T. Heidmann. 2007. Placental syncytins: genetic disjunction between the fusogenic and immunosuppressive activity of retroviral envelope proteins. Proceedings of the National Academy of Sciences, USA 104:20534–20539.
Mitchell, R. S., B. F. Beitzel, A. R. Schroder, P. Shinn, H. Chen, C. C. Berry, J. R. Ecker, and F. D. Bushman. 2004. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biology 2:E234.
Nelson., P. N., P. R. Cargenie, J. Martin, H. Davari Ejtehadi, P. Hooley, D. Roden, S. Rowland-Jones, P. Warren, J. Astley, J., and P. G. Murray. 2003. Demystified . . . human endogenous retroviruses. Journal of Clinical Pathology: Molecular Pathology 56:11–18.
Nyswaner, K. M., M. A. Checkley, M. Yi, R. M. Stephens, and D. J. Garfinkel. 2008. Chromatin-associated genes protect the yeast genome from Ty1 insertional mutagenesis. Genetics 178:197–214.
Okulicz, W. C., and C. I. Ace. 2003. Temporal regulation of gene expression during the expected window of receptivity in rhesus monkey endometrium. Biology of Reproduction 69:1593–1599.
Ono, M., M. Kawakami, and H. Ushikubo. 1987. Stimulation of expression of the human endogenous retrovirus genome by female steroid hormones in human breast cancer cell line T47D. Journal of Virology 61:2059–2062.
Peaston A. E., A. V. Evsikon, J. H. Graber, W. N. de Vries, A. E. Holbrook, D. Solter, and B. B. Knowles. 2004. Retrotransposons regulate host genes in mouse oocytes and preimplantation embryos. Developmental Cell 7:597–606.
Prudhomme, S., G. Oriol, and F. Mallet. 2004. A retroviral promoter and a cellular enhancer define a bipartite element which controls env ERVWE1 placental expression. Journal of Virology 78:12157–12168.
Reiss, D., and D. L. Mager. 2007. Stochastic epigenetic silencing of retrotransposons: Does stability come with age?Gene 390:130–135.
Rinckel, L. A., and D. J. Garfinkel. 1996. Influences of histone stoichiometry on the target site preference of retrotransposons Ty1 and Ty2 in Saccharomyces cerevisiae. Genetics 142:761–776.
Sanford, J. 2008. Genetic entropy and the mystery of the genome, 3rd ed., pp. 38–39. Waterloo, New York: FMS Publications.
Schulz, W. A., C. Seinhoff, and A. R. Flori. 2006. Methylation of endogenous retroelements in health and disease.Current Topics in Microbiology and Immunology 310:211–250.
Shapiro, J. A. 2005. Retrotransposons and regulatory suites. Bioessays 27:122–125.
Spence, S. E., D. J. Gilbert, D. A. Swing, N. G. Copeland, and N. A. Jenkins. 1989. Spontaneous germ line virus infection and retroviral insertional mutagenesis in eighteen transgenic Srev lines of mice. Molecular and Cellular Biology 9:177–184.
Spencer, T. E., M. Mura, C. A. Gray, P. J. Griebel, and M. Palmarini. 2003. Receptor usage and fetal expression of ovine endogenous betaretroviruses: implications for coevolution of endogenous and exogenous retroviruses. Journal of Virology 77:749–753.
Turner, G., M. Barbuscu, M. Su, M. I. Jensen-Seaman, K. K. Kidd, and J. Lenz. 2001. Insertional polymorphisms of full-length endogenous retroviruses in humans. Current Biology 11:1531–1535.
van de Lagemaat, L. N., P. Medstrand, and D. L. Mager. 2006. Multiple effects govern endogenous retrovirus survival patterns in human gene introns. Genome Biology 7:R86.
Wadsworth, J. D. F., A. F. Hill, J. A. Beck, and J. Collinge. 2003. Molecular and clinical classification of human prion disease. British Medical Bulletin 66:241–254.
Weiss, R. A. 2006. The discovery of endogenous retroviruses. Retrovirology 3:67.
Weiss, R. A., R. R. Friis, E. Katz, and P. K. Vogt. 1971. Induction of avian tumor viruses in normal cells by physical and chemical carcinogens. Virology 46:920–938.
Yu, C., K. Shen, M. Lin, P. Chen, C. Lin, G. Chang, and H. Chen. 2002. GCMa regulates the syncytin-mediated trophoblast fusion. The Journal of Biological Chemistry 277: 50062–50068.
No comments:
Post a Comment